> Biology Group. 2021 May 26;2(5):383-391. doi: 10.37871/jbres1246.
-
Subject area(s):
- Biochemistry
- Molecular Biology
- Bioinformatics
- Bioengineering
- Biotechnology
- Biology
- Virology
![]()
 |
|
![]() |
|
![]() |
|
![]()
Effect of Positional Isomerism on Some Alcohol Based Drug towards Anti-Viral Activity against SARS-Cov-2: A Molecular Modeling Based Investigation
Mahendiali Palsaniya1#, Bansari Patel1#, Nibedita Panigrahi2, Daffodil Mohanta3, Sonali Priyadarshini Parida3, Dhruvin Kumar Patel1, Mriganka Das1* and Bidyut Kumar Kundu3*
2Department of Chemistry, Central University of Jharkhand, Jharkhand, India
3School of Applied Sciences, Centurion University of Technology and Management, Odisha, India
#These authors contributed equally
Abstract
The severe acute respiratory syndrome coronavirus 2, better known as COVID-19, has become a major health concern worldwide. It has challenged the global healthcare sector like anything. It appeared in Wuhan, China, around November 2019, had spread to almost 187 countries due to its highly contagious nature. Quarantine, isolation, mask, and other precautionary measures remain the sole obliging strategy to decline the person-to-person transmissions. Amidst the pandemic, drug repurposing by identifying therapeutically potent molecule from the collection of pre-existing molecules by molecular docking and DFT methods are certainly fast and handy. Herein, this paper is dealing with 5 hydroxy based drugs such as 5-isopropyl-2-methylphenol (Carvacrol), 3-isopropyl-6-methylbenzene-1,2-diol, 2-isopropyl-5-methylbenzene-1,4-diol, 5-isopropyl-2-methylbenzene-1,3-diol, 2-isopropyl-5-methylbenzene-1,3-diol to discover the new possible COVID-19 inhibitors. The proteases PDB, e.g., 5r7y is used as hosts to calculate the interactions with hydroxy-based drugs as guests. Our research shows that 5-isopropyl-2-methylbenzene-1,3-diol is the most active, having binding energy –6.46 kcal/mol against 5r7y of SARS-CoV-2. Hence it is assumed that increasing number of alcohol group make the system more preferable towards SARS-CoV-2 protease protein 5r7y. It was also observed that relative binding energy among these alcohol-based drugs is further tuned by their positional isomerism property.
Introduction
Coronavirus disease 2019 or COVID-19 has created devastation in humankind throughout the world. The virus, seemingly unstoppable, highly contagious, and infectious, is in an exponential growth phase worldwide with numbers increasing day by day. There have been 14,81,28,030 confirmed cases of COVID–19, including 31,24,905 deaths worldwide as of 28th April 2021 reported to the World Health Organization (WHO). COVID–19 has been accepted as a pandemic by the World Health Organization (WHO) on 11th March 2020. Almost all the continents and countries have suffered from this pandemic hugely. Initially, COVID- 19 started in the Wuhan city of Hubei province in China in November 2019. It was spreading as an unknown pneumonia-type virus having many similar symptoms with Severe Acute Respiratory Syndrome (SARS) in 2003, Middle East Respiratory Syndrome (MERS) in 2013, and other influenza viruses [1]. But COVID–19 is acknowledged as much more virulent than MERS and SARS [2]. The most common symptoms of COVID-19 patients are fever, fatigue, dry cough, myalgia, and dyspnea [3]. Sputum production, headache, hemoptysis, and diarrhea are the other indications of infected patients [4]. The major routes of human to human transmission is sneezing droplets and air [5].
Covid-19 virus transfers from an infected person to a normal person via aerosol transmission also. Exhalation, coughing, sneezing and talking to an infected person can spread it as SARS-CoV-2 can be suspended in air for hours and travels via air [6].
Coronaviruses are categorized as a member of the subfamily Coronavirinae, family Coronaviridae, and order Nidovirales. Four major genera of this subfamily are Alpha – CoVs, Beta – CoVs, Gamma – CoVs, and Delta-CoVs [7]. Alpha and Beta CoVs are mainly known to infect mammals generating from primary host bats and rodents [8]. The diameter of enveloped viral component COVID-19 virus is 60-160nm though it primarily occurs in the pleomorphic form [9].
Coronaviruses are virions having crown-like projections of glycoproteins protruding outside their near-spherical shape [1,4]. Coronavirus has an ssRNA genome enveloped through the folded structure [10]. These ssRNAs possess a 5’-cap arrangement and 3’ – poly-A tail, which interacts with nucleoproteins [7]. Almost all CoVs share resemblances in genome organization and expression. The genome of CoV encodes 4 key structural proteins: the Spike protein(S), Nucleocapsid protein (N), Membrane protein (M), and the Envelope protein (E [11,12]. COVID-19 shares 50%, 79%, and 96% genetic resemblance to MERS-CoV, SARS-CoV, and other bat SARS-related viruses, respectively [13,14]. SARS-CoV-1 and SARS-CoV-2 are also noticed to share Angiotensin-Converting Enzyme II (ACE2) as their receptor [15].
Scientists are tirelessly working for the advancement of immunization and medications to deal with COVID-19. Phyto compounds are also under research. Bioactive Phyto-intensifies, which were usually utilized, has now developed as vital medications to treat viral infection. When the COVID-19 virus enters the host’s non-specific innate immune responses and then antigen adaptation by mediated B cells and regulatory T cells. On the first meeting with SARS- CoV-2 memory is created by B and T cells. This is known as protecting immunity. It stimulates a quicker and stronger response against the COVID-19 virus on successive encounters [16].
The biological mixture-based therapeutic procedure can be focused on, which are composed of organic compounds having hydroxyl or keto groups. Previous studies show that several fundamental mixtures, where the major counterpart is hydroxyl functional group-based organic ligands, have successfully inhibited the attack of microbes. A couple of fundamental organic ligands have been viable against RNA and DNA infections, for example, avian flu infection [17], Herpes Simplex Infection Type 1 (HSV-1) & Type 2 (HSV-2), dengue infection type 2, Junín infection, flu infection adenovirus type 3, poliovirus, and coxsackievirus [18]. As there is an inadequate number of medications available for treating the viral ailment, a specific understanding of drug development is the obvious need of this hour. For evaluating the therapeutic potential of a specific drug, it is crucial to comprehend the association between the ligands and the protein at the sub-atomic level. An underlying in-silico research incorporating various methods impressively can reduce the time needed for this drug discovery process [19,20]. It is easy to find out possible results about protein-ligand or host-guest interactions through computational studies by analyzing some specific parameters (e.g. binding energy, number of hydrogen bonds, hydrophobic bonds, bandgap, etc.), that will assist further in targeted drug designing.
This article has selected the SARS-CoV-2 principle protease as the host for the situation with some hydroxy-based organic ligands. [21,22] The protease receptor proteins PDB ID: 5r7y had been chosen. In the current experiment, molecular docking and the applied Density Functional Theory (DFT) technique have been used to evaluate hydroxy-based drugs. Using molecular docking, hydrogen bonding and hydrophobic interactions have been tested properly in the stable collaboration between the protein and ligand.
Materials and Methods
Selection and preparation of host protein structure
The protease subunit of SARS-CoV-2 proteins like PDB ID: 5r7y is specifically selected as the target with selected ligand molecules. The reason behind the selection of this protease subunit is because of its vital role in the viral effect. From the RCSB protein data bank, both protease subunits were downloaded in PDB format. For visualizing the 3D structure and removal of water molecules and unwanted species bound to the host PDB, Pymol was used. After the successful creation of the PDB host from Pymol, Autodock software was used for modifying various charges and energies correlated with the PDB of hosts. For further use PDB was converted into pdbqt format with the help of the same software.
Selection and preparation of ligands
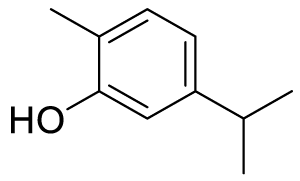
Here we take five hydroxy compounds as a ligand viz 5-isopropyl-2-methyl phenol, 3-isopropyl-6-methylbenzene-1,2-diol, 2-isopropyl-5-methylbenzene-1,4-diol, 5-isopropyl-2-methylbenzene-1,3-diol, and 2-isopropyl-5-methylbenzene-1,3-diol. Among these ligands, 5-isopropyl-2-methyl phenol (Carvacrol) is experimentally known for its antiviral, and/or anti-bacterial activities. These are selected as guests against the SARS-CoV-2 protease-based host viz; 5r7y. Now, ChemDraw3D along with MM2 performance has been used to generate each structure of ligand for finding out detailed information about their exact chemical composition. The exported PDB of the ligands from ChemDraw was further used to calculate the geometry optimized structure through Density Functional Theory (DFT). These ligands were screened at Auto dock software which converts PDB files into pdbqt format. This process involves detecting torsion root, adjusting torsion angle, assigning charges, and changing them to pdbqt format for their further use during molecular docking [23-26].
Theoretical calculations of ligand structures
Density Functional Theory (DFT) gives us an idea of the solid-state structure of a molecule using quantum formulations [27]. Another field of DFT is to state chemical behavior by using electron density calculation. To achieve the geometry optimized structures, the Gaussian09 program having B3LYP basis set and 6-31G(+)d,p functional have been used for each ligand. Furthermore, different parameters have been elucidated via DFT calculations, which includes total energy, molecular dipole moment, Lowest Unoccupied Molecular Orbital (LUMO) and Highest Occupied Molecular Orbital (HOMO) energies, bandgap (ΔE), absolute hardness (ɳ), a fraction of electrons transfer (ΔN), and electronegativity (χ) [24].
Molecular docking
In-silico technique anticipates suitable interconnection between protein molecules of hosts and small guests (viz. ligand) based on their geometry and structure [28]. In this report, molecular docking is performed by using certain software and servers such as Autodock, CCDC Gold, PLIP, Open Bable GUI, Chimera, and Protein. Plus.
In Autodock, accuracy and speed are maintained throughout the steps. The protein and the ligand molecules were selected with a little adjustment of Kollman and residual charges. Apart from that, the structure is dehydrated and heteroatom(s) were deleted wherever applicable. Furthermore, torsion tree grid and spacing were set followed by running ‘auto grid’ and ‘auto dock’ programs to get the host-guest interaction-based docked results in the form of binding energy and inhibition constant [29].
The best dock PDB file obtained in Autodock were selected and converted to pdbqt format in the open babel tool. The cavity site recognition is known to be a preliminary step for protein binding site recognition was done through protein plus server and the 3D structure of molecules was observed through the PLIP server.
Besides, CCDC Gold work proceeded by choosing the HERMES tool, in which protein in PDB format is loaded followed by defining binding sites. Mol2 file of ligand prepared from CCDC Mercury was selected by using ligand flexibility, GA setting, scoring function, and all other required functions to run a program, which after completion gives docking solutions/poses.
Chimera is an extensible program for instinctive portrayal and examination of nuclear developments and related data, including thickness maps, supramolecular social occasions, gathering game plans, docking results, bearings, and conformational outfits. Structural analysis is done using this, all the residues were also named using the name specifier option. At last interaction poses and sessions were saved for further use.
Results and Discussion
Molecular docking
The docking technique is an in silico technique, where the measurement of binding energy parameters and scores are used to predict how a protein interacts with ligands [30,31]. To determine potential antiviral activity, all the ligands are docked with host SARS-COV-2 proteins. So, the docking result of 5-isopropyl-2-methylbenzene-1,3-diol with 5r7y gives the lowest value of binding energy (-6.46 kcal/mol) during complexation and it is a leading score compare to other docked results. The binding energy result with other ligands are 2-isopropyl-5-methylbenzene-1,4-diol (-6.09 kcal/mol), 5-isopropyl-2-methyl phenol (-5.78 kcal/mol), 2-isopropyl-5-methylbenzene-1,3-diol (-5.29 kcal/mol), and 3-isopropyl-6-methylbenzene-1,2-diol (-5.11 kcal/mol) are lower than binding energy of 5-isopropyl-2-methylbenzene-1,3-diol. Five hydrogen bonding are shown with amino acids ASP295A (H-donor), GLN299A (H-donor), GLN299A (H-donor), MET6A (H-donor), PHE8A (H-donor) and four hydrophobic interaction with amino acid residues PHE8A, ASP295A, ARG298A, GLN299A are laid out in complexation with the host 5r7y. The hydrogen bonding and hydrophobic interaction between other ligands and protein 5r7y are briefed in table 2.
In the formation process of protein-ligand complexes, hydrogen bonding plays a vital role in determining its specificity and affinity of complexes. Hydrogen bonding and hydrophobic interactions play an important role in giving shape and stabilizing the docked complex. Apart from describing binding energy, a comparison of inhibition constant (Ki) can additionally provide information about inhibitor potential. Here 5-isopropyl-2-methylbenzene-1,3-diol with 5r7y indicates excellent Ki value of 18.47μM in comparison with 2-isopropyl-5-methylbenzene-1,4-diol (34.35μM), 5-isopropyl-2-methyl phenol (57.65μM), 2-isopropyl-5-methylbenzene-1,3-diol (132.28μM), and 3-isopropyl-6-methylbenzene-1,2-diol (181.13μM). The hydrogen bonding and hydrophobic interaction between the selected ligands and the host 5r7y are summarized in table 1. 2D binding poses are calculated using protein plus server and depicted in figure 1. From this calculation, it is clear that when two -OH group is placed in meta and para position, then a severe interaction is happening with amino acid residues of the proteins.

| Table 1: Ligands structure in two and three dimensions. | ||
| Compound Name | Chemical Structure | |
| 2D | 3D | |
| 5-isopropyl-2-methylphenol (Carvacrol) (C1) |
 |
 |
| 3-isopropyl-6-methylbenzene-1,2-diol (C2) |
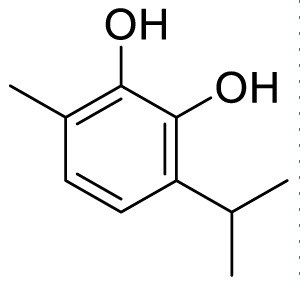 |
 |
| 2-isopropyl-5-methylbenzene-1,4-diol (C3) | 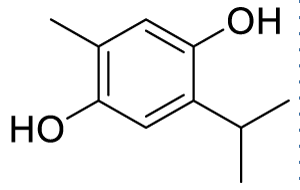 |
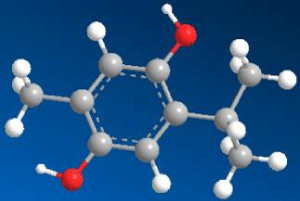 |
| 5-isopropyl-2-methylbenzene-1,3-diol (C4) | 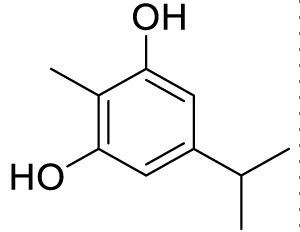 |
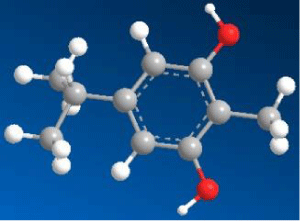 |
| 2-isopropyl-5-methylbenzene-1,3-diol (C5) |  |
 |
| Table 2: Obtained best scores from docking of ligand 5-isopropyl-2-methylphenol, 3-isopropyl-6-methylbenzene-1,2-diol, 2-isopropyl-5-methylbenzene-1,4-diol, 5-isopropyl-2-methylbenzene-1,3-diol, and 2-isopropyl-5-methylbenzene-1,3-diol with host protein 5r7y of SARS-CoV-2 virus. | ||||||||
| Ligand | Protein of Host | RMSD* | Binding energy (Kcal/mol) |
Inhibition constant (Ki) |
No of H-bonds (drug-protein) | Amino acid involved in the interaction | No of Hydro phobic bond |
Amino acid residue involved |
| 5-isopropyl-2-methylphenol (C1) |
5r7y | 0.80 | -5.78 | 57.65μM | 2 | ASP295A GLN299A |
6 | ARG298A PHE8A PHE8A PHE8A PHE291A ASP295A |
| 3-isopropyl-6-methylbenzene-1,2-diol (C2) |
5r7y | 0.29 | -5.11 | 181.13μM | 3 | MET6A MET6A GLN127A |
4 | PHE8A PHE291A ASP295A GLN299A |
| 2-isopropyl-5-methylbenzene-1,4-diol (C3) |
5r7y | 0.21 | -6.09 | 34.35μM | 4 | ALA7A ASP295A GLN299A GLN299A |
3 | MET6A ASP295A GLN299A |
| 5-isopropyl-2-methylbenzene-1,3-diol (C4) |
5r7y | 0.22 | -6.46 | 18.47μM | 5 | MET6A PHE8A ASP295A GLN299A GLN299A |
4 | PHE8A ASP295A ARG298A GLN299A |
| 2-isopropyl-5-methylbenzene-1,3-diol (C5) |
5r7y | 1.39 | -5.29 | 132.28μM | 4 | MET6A ASP295A GLN299A GLN299A |
4 | MET6A GLN299A ASP295A ARG298A |
| *RMSD value of the best-docked conformation. | ||||||||
Furthermore, the docked ligands with hydrophobic interactions that are selected for identifying suitable binding pocket(s) around the ligands are summarized via Protein. Plus server. This hydrophobic interaction between the ligands and the host protein gives the information about the stability of the docked complex. The binding pocket of each ligand is shown in figure 2, where different colors have been used for better visualization and easy investigation. PLIP outcomes are summarized in figure 3, which provide not only the 3D plot but also the data related to hydrophobic interaction, an amino acid involved in the interaction, and residue of that specific amino acid. Apart from that, hydrogen bonding and hydrophobic interactions play an important role in giving shape and stabilizing the docking complex. From the results, it can be concluded that the number of hydrogen bonds, as well as hydrophobic interactions, are proportional to the activity of the corresponding ligand.

PLIP results are summarized in figure 3, which gives not only the 3D plot but also the data related to hydrophobic interaction, an amino acid involved in the interaction, and residue of that specific amino acid. Apart from that, hydrogen bonding and hydrophobic interactions play an important role in giving shape and stabilizing the docking complex. From the results, it can be concluded that the number of hydrogen bonds, as well as hydrophobic interactions, are proportional to the activity of the corresponding ligand.

CCDC Gold has been used to authenticate the Autodock results by choosing all essential frameworks like torsion angle distribution, protonated ligand, rotatable bond, flexibility, and GA setting to run the process. From these, we get binding sites accompanying several solutions, which are presented in figure 4. The results are investigated by analogizing the bond distance from the functional groups of ligands with the amino acid residue of certain proteases of the host SARS-CoV-2. For example, 3-isopropyl-6-methylbenzene-1,2-diol interacts with the lowest bond distance of 1.963Å with GLN189. An amino residue of host 5r7y. The bond distance of the host-guest plays a vital role in binding interaction and it is observed that the activity of the host-guest molecule is more if the bond distance is less between host-guest. By using the chimera tool, the 3D structure of the host-guest complex of the best-docked analog was elucidated to determine the active binding sites of the ligand with protein (Figure 4).

From the all data as described above, it is clear compounds containing two -OH group bind more effectively with 5r7y protein than the compound which has a single -OH group. Not only number of -OH group is primary attention towards high binding but also the position of -OH group is a major concern here. Meta and para diol compounds are more effective than ortho diol. This is because of the interactions of different amino acids from different regions. It was further observed that compound 4 (C4) meta diol is most effective. This is might be because of less steric repulsion between two meta-oriented -OH groups. There is a tertiary butyl group presents in between two meta-oriented -OH groups. Thus, there is a severe steric repulsion. For this reason compound, 3 becomes less effective than compound 4.
Evaluation of drug-likeness
5-isopropyl-2-methyl phenol (C1), 3-isopropyl-6-methylbenzene-1,2-diol (C2), 2-isopropyl-5-methylbenzene-1,4-diol (C3), 5-isopropyl-2-methylbenzene-1,3-diol (C4), and 2-isopropyl-5-methylbenzene-1,3-diol (C5) have respectively the following molecular weights (150.22, 166.22, 166.22, 166.22, 166.22) g/mol, all five molecule have molecular weight ≤ 500 g/mol which follow the Lipinski’s rule. The 5-isopropyl-2-methyl phenol molecule has been found to have topological polar surface (TPSA) as 20.23 Å2, while 3-isopropyl-6-methylbenzene-1,2-diol, 2-isopropyl-5-methylbenzene-1,4-diol, 5-isopropyl-2-methylbenzene-1,3-diol, and 2-isopropyl-5-methylbenzene-1,3-diol molecules were found to have 40.46 Å2. According to the rule, the lowest TPSA values always produce good results; therefore, we noted that selected molecules are better behaved than the co-crystallized ligand. [32] The (LogP) values of C1, C2, C3, C4, and C5 were found in the range of 2.82, 2.35, 2.39, 2.31, 2.34, respectively. Predicted LogP values depict that these molecules can be absorbed in the body. All five molecules present a number of hydrogen bond donors: ≤5, a number of hydrogen bond acceptors ≤10, and also molar refractivity values between 48.01, 50.03, 50.03, 50.03, 50.03, respectively. The hydrogen bonding and a molar refractivity calculation showed that these molecules validate the five Lipinski’s rule. These molecules also follow the veber’s rule which denotes the oral bioavailability of drug molecules. Since in some CAD molecules cannot be synthesized, synthetic accessibility (SA) is a key aspect of drug designing. The 5-isopropyl-2-methylphenol, 3-isopropyl-6-methylbenzene-1,2-diol, 2-isopropyl-5-methylbenzene-1,4-diol, 5-isopropyl-2-methylbenzene-1,3-diol, and 2-isopropyl-5-methylbenzene-1,3-diol molecules were found to have SA score 1.00, 1.04, 1.05, 1.08, 1.07, respectively. According to SA method molecules having score 1 is easy to synthesize it; on the other hand, score 10 represents difficulties to synthesize the molecules. All five molecules having less than 10 score, so they can be easily synthesized. The results of the ADME calculations are listed in the table 3.
| Table 3: Results of phytochemical molecules drug likeness properties. | |||||
| Drug Likeness: Ligands | C1 | C2 | C3 | C4 | C5 |
| Molecular Weight g/mol | 150.22 | 166.22 | 166.22 | 166.22 | 166.22 |
| Consensus Log P0/W | 2.82 | 2.35 | 2.39 | 2.31 | 2.34 |
| Num. H-bond acceptors | 1 | 2 | 2 | 2 | 2 |
| Num. H-bond donors | 1 | 2 | 2 | 2 | 2 |
| Molar Refractivity | 48.01 | 50.03 | 50.03 | 50.03 | 50.03 |
| Lipinski | Yes | Yes | Yes | Yes | Yes |
| Veber | Yes | Yes | Yes | Yes | Yes |
| Bioavailability | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
| Synthetic accessibility (SA) | 1.00 | 1.04 | 1.05 | 1.08 | 1.07 |
| TPSA (Å2) | 20.23 | 40.46 | 40.46 | 40.46 | 40.46 |
| No of rotatable bonds | 1 | 1 | 1 | 1 | 1 |
| Solubility | 1.46e-01 | 5.91e-01 | 5.91e-01 | 5.91e-01 | 5.91e-01 |
Concepts of DFT
Initially, the geometry optimized solid-state structures of the keto-based ligands are calculated through Density Functional Theory (DFT). Also, molecular orbital energies like HOMO (EHOMO) and LUMO (ELUMO) were calculated for those ligands to check their tendency to donate and/or accept electrons towards protease hosts viz. 5r7y. The electron density in different regions of the molecule at HOMO and LUMO is generated and visualized in figure 5. Furthermore, the HOMO energy (EHOMO) and LUMO energy (ELUMO) values of the selected natural drugs are summarized in table 2. Besides, the electron density maps of molecular orbitals of the chosen ligands are shown in figure 5 and the energy or bandgap (ΔE) between two molecular orbitals of the ligand is calculated with the formula: ΔE = ELUMO - EHOMO.

Further, the ionization potential and electron affinity can be calculated by using formulas I= -EHOMO and A= -ELUMO, respectively. We also derived the values of electronegativity (χ), absolute hardness (η) and a fraction of electron transfer (ΔN) by using the formula χ = (I+A)/2, η = (I-A)/2 and ΔN = χFe-χinh/2(ηFe+ηinh), respectively. We considered the theoretical value of χFe = 7 eV and ηFe =0 eV to calculate ΔN value.
Guest compounds (C1, C2, C3, C4, and C5) that are taken for this study give molecular dipole moment of 1.5167 Debye, 2.5757 Debye, 0.1618 Debye, 2.2820 Debye, and 1.6174 Debye, respectively (Table 4).
| Table 4: DFT calculated parameters to obtain the value of dipole moment, electronegativity (χ), absolute hardness (η), and a fraction of electron transfer (ΔN). | |||||||
| Ligand | EHOMO (eV) |
ELUMO (eV) |
Band Gap (ΔE in eV) | ELUMO(Fe) - EHOMO (inh in eV) |
Electro negativity (χ) |
Absolute Hardness (ɳ) |
fraction of electrons transfer (ΔN) |
| C1 | -6.0481 | -0.2312 | 5.8169 | 6.1781 | 3.13965 | 2.90845 | 0.6636438 |
| C2 | -5.8618 | -0.4545 | 5.4073 | 5.9918 | 3.15815 | 2.70365 | 0.7104932 |
| C3 | -5.5602 | -0.3087 | 5.2515 | 5.6902 | 2.93445 | 2.62575 | 0.7741692 |
| C4 | -5.9241 | -0.5676 | 5.3565 | 6.0541 | 3.24585 | 2.67825 | 0.7008587 |
| C5 | -5.9565 | -0.2369 | 5.7196 | 6.0865 | 3.0967 | 2.8598 | 0.6824428 |
Conclusion
COVID-19 should be restrained to curb further spread and mortality as soon as possible. The natural organic ligands found in several plants with potential antimicrobial or antifungal actions have been proposed for this study. Thus, these ligands can play a leading position in deterring the recurrence of the virus in the gripping system and thus stopping additional devastation. Searching for a desirable binding position inside the host protein(s) is very significant to fix its viral activities. Therefore, molecular docking simulationion is of great interest to recent research to select site-specific drugs with the targeted role. With the incorporation of the DFT approach, this research has contributed to a better understanding of the chemical nature of hydroxy-based natural drugs by defining the electron density of molecules. At a glimpse, the outcomes of this in silicon techniques using virtual examination can be very useful to find some phyto-compounds or natural drugs suitable for the treatment of COVID-19 viral infection.
References
- Tu H, Tu S, Gao S, Shao A, Sheng J. Current epidemiological and clinical features of COVID-19; a global perspective from China. J Infect. 2020 Jul;81(1):1-9. doi: 10.1016/j.jinf.2020.04.011. Epub 2020 Apr 18. PMID: 32315723; PMCID: PMC7166041.
- Singh A, Shaikh A, Singh R, Singh AK. COVID-19: From bench to bed side. Diabetes Metab Syndr. Clin Res Rev. 2020; 14:277-281. doi:10.1016/j.dsx.2020.04.011.
- Shah V, Bhaliya J, Shah D. In Silico Study of 1,5-Bis(4-Hydroxy-3-Methoxyphenyl)-1,4-Pentadiene-3-One (Deketene Curcumin) on Crystallized Protein Structures of SARS-CoV-2. ChemRxiv. 2020;19:1-15. doi:10.26434/chemrxiv.12377000.
- Hu Y, Liang W, Liu L, Li L. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med. 2020;18: 1708-1728. doi:10.1056/NEJMoa2002032.
- Song F. Emerging 2019 Novel Coronavirus (2019-nCoV) Pneumonia. Radiology. 2020;295:210–217. doi:10.1148/radiol.2020200274.
- Priyanka, Choudhary OP, Singh I, Patra G. Aerosol transmission of SARS-CoV-2: The unresolved paradox. Travel Medicine and Infectious Disease 2020;37:101869. doi:10.1016/j.tmaid.2020.101869.
- Chang L, Yan Y, Wang L. Coronavirus Disease 2019: Coronaviruses and Blood Safety. Transfus Med Rev. 2020;34: 75-80. doi:10.1016/j.tmrv.2020.02.003.
- Woo PCY. Discovery of Seven Novel Mammalian and Avian Coronaviruses in the Genus Deltacoronavirus Supports Bat Coronaviruses as the Gene Source of Alphacoronavirus and Betacoronavirus and Avian Coronaviruses as the Gene Source of Gammacoronavirus and Deltacoronavi. J Virol. 2012;86:3995-4008. doi:10.1128/JVI.06540-11.
- Kang S. Recent progress in understanding 2019 novel coronavirus (SARS-CoV-2) associated with human respiratory disease: detection, mechanisms and treatment. Int J Antimicrob Agents 2020;55:105950. doi:10.1016/j.ijantimicag.2020.105950.
- Yang, H, Rao MB. Drug Design Targeting the Main Protease, the Achilles Heel of Coronaviruses. Current Pharmaceutical Design 2006;12:4573-4590. doi:10.2174/138161206779010369.
- Masters PS. The Molecular Biology of Coronaviruses. Advances in Virus Research. 2006;65:193-292. doi:10.1016/S0065-3527(06)66005-3.
- Schoeman D, Fielding BC. Coronavirus envelope protein: Current knowledge. Virol J. 2019;16:69. doi:10.1186/s12985-019-1182-0.
- Li W. Bats Are Natural Reservoirs of SARS-Like Coronaviruses. Science. 2005;310:676-679. doi:10.1126/science.1118391.
- Gralinski LE, Menachery VD. Return of the Coronavirus: 2019-nCoV. Viruses 2020;12. doi:10.3390/v12020135.
- Tang, X. On the origin and continuing evolution of SARS-CoV-2. Natl Sci Rev. 2020;7:1012–1023. doi:10.1093/nsr/nwaa036.
- Priyanka, Choudhary OP, Singh I. Protective immunity against COVID-19: Unravelling the evidences for humoral vs. cellular components. Travel Medicine and Infectious Disease. 2021;39:101911. doi:10.1016/j.tmaid.2020.101911.
- Pourghanbari G, Nili H, Moattari A, Mohammadi A, Iraji A. Antiviral activity of the oseltamivir and Melissa officinalis L. essential oil against avian influenza A virus (H9N2). Virus Disease 2016;27:170-178. doi:10.1007/s13337-016-0321-0.
- Tariq S. A comprehensive review of the antibacterial, antifungal and antiviral potential of essential oils and their chemical constituents against drug-resistant microbial pathogens. Microb Pathog. 2019;134:103580. doi:10.1016/j.micpath.2019.103580.
- Terstappen GC, Reggiani A. <em>In silico</em> research in drug discovery. Trends Pharmacol. Sci. 2001;22:23-26. doi:10.1016/S0165-6147(00)01584-4.
- Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br J Pharmacol. 2007;152:9-20. doi:10.1038/sj.bjp.0707305.
- Lan J. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581: 215-220. doi:10.1038/s41586-020-2180-5.
- Zhang L. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409-412. doi:10.1126/science.abb3405.
- Kumar A. Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. J Biomol Struct Dyn. 2020;1-11. doi:10.1080/07391102.2020.1772112.
- Das M. Targeted synthesis of cadmium(II) Schiff base complexes towards corrosion inhibition on mild steel. RSC Adv. 2017;7:48569-48585. doi:10.1039/c7ra08633d.
- Das M. Enhanced pseudo-halide promoted corrosion inhibition by biologically active zinc(II) Schiff base complexes. Chem Eng J. 2019;357:447-457. doi:10.1016/j.cej.2018.09.150.
- O’Boyle NM. Open Babel: An open chemical toolbox. J Cheminform. 2011;3:33. doi:10.1186/1758-2946-3-33.
- Geerlings P. Conceptual density functional theory: status, prospects, issues. Theor Chem Acc. 2020;139:36. doi:10.1007/s00214-020-2546-7.
- Leach AR, Shoichet BK, Peishoff CE. Prediction of Protein−Ligand Interactions. Docking and Scoring: Successes and Gaps. J Med Chem. 2006;49:5851-5855. doi:10.1021/jm060999m.
- Vieira TF, Sousa SF. Comparing AutoDock and Vina in Ligand/Decoy Discrimination for Virtual Screening. Applied Sciences 2019;9. doi:10.3390/app9214538.
- Elfiky AA, Ismail AM, Elshemey WM. Recognition of gluconeogenic enzymes; Icl1, Fbp1, and Mdh2 by Gid4 ligase: A molecular docking study. J Mol Recognit. 2020;33:e2831. doi:10.1002/jmr.2831.
- Elfiky AA. Novel guanosine derivatives against Zika virus polymerase in silico. J Med Virol. 2020;92:11–16. doi:10.1002/jmv.25573.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3-26. doi:10.1016/S0169-409X(00)00129-0.




Content Alerts
SignUp to our
Content alerts.
 This work is licensed under a Creative Commons Attribution 4.0 International License.
This work is licensed under a Creative Commons Attribution 4.0 International License.
